|
|

| Prion
diseases are fatal neurodegenerative diseases that include Creutzfeldt-Jacob
disease (CJD) in humans, mad cow disease
in cattle and scrapie in sheep. The infectious
agent in transmissible spongiform encephalopathies is a proteinous infectious
particle or ‘prion’, which is devoid of nucleic acid.
It is believed that these spongiform encephalopathies are caused
by the accumulation of an abnormally folded isoform of the cellular
prion protein (PrPC). This misfolded protein is rich in beta-sheet
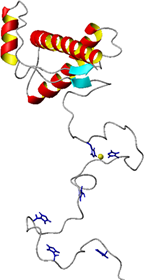
and is designated the scrapie isoform, (PrPSc). The normal physiological function of the prion protein is yet to be determined. However, the ability of PrPC to bind Cu2+ in vivo and in vitro suggests a role in copper homeostasis (Viles, 1999). Indeed, elevated copper levels promote endocytosis of PrP suggesting that PrP could transport copper into the cell. |
|
Structurally, PrPC contains two distinct regions. In the absence of Cu2+, the N-terminal domain, residues 23-120, is unstructured and has a high degree of main-chain flexibility in the absence of copper (Viles, 2001). In contrast, the C-terminal domain, residues 121-231, is largely a-helical. Residues 60-91 consist of an octapeptide sequence, PHGGGWGQ, which is repeated four times. It is this unstructured region that binds four Cu2+ ions cooperatively (Viles, 1999).
|
|
The
Viles Group is interested in the Prion protein in a number of
areas. In general these involve: 1. Investigating the metal binding properties of various regions of the protein and, 2. Investigating the folding and misfolding properties of the protein, and the role that metals play in this process. We use many spectroscopic techniques, including multi-dimensional NMR, EPR, CD, fluorescence and UV/Vis absorption spectroscopy and X-ray Spectroscopy (EXAFS). |
|